


In vivo dual cross-linking chromatin immunoprecipitation: detecting chromatin proteins not directly bound to DNA (PROT29)

Antonio Porro and Giovanni Perini
University of Bologna
Department of Biology
University of Bologna
Via Francesco Selmi 3
40126 Bologna, Italy
Email feedback to:
[email protected]
Introduction
Cross-linking Chromatin Immunoprecipitation (ChIP) has become a popular method to detect the in vivo binding of proteins to DNA. In general the protein/DNA complex is fixed with formaldehyde, a cross-linking agent that, because of its short spacer arm (about 2 A), generates reversible covalent links, mainly between proteins and DNA. Subsequently, chromatin is fragmented by sonication and DNA protein complexes are selectively immunoprecipitated with antibodies raised against proteins of interest. Covalent links are reversed and proteins are removed from DNA through phenol/chloroform extraction. Following DNA purification, specific DNA regions supposed to be engaged with proteins of interest are analyzed by semi-quantitative PCR or real time PCR. Although very powerful, this method may present some limits when applied to nuclear proteins that, although being tightly associated with chromatin DNA, are not directly bound to it. For example, recent studies have shown that c-Myc can repress transcription of the p21CIP/WAF gene without binding DNA directly but through interaction with different transcription factors such as Sp1 and Miz-1 that recognize specific DNA sequences in the p21CIP/WAF promoter (Gartel et al., 2001 and Wu e al., 2003). N-Myc, another member of the Myc family, is expressed essentially during development of the nervous system and in some rare pediatric tumors such as neuroblastoma, medulloblastoma and rhabdomyosarcoma. We have recently defined some parts of the network of genes that N-Myc can regulate in human neuroblastoma (Perini et al., 2005). Many target genes are positively regulated by N-Myc; nonetheless, we have found that several genes are also negatively regulated by N-Myc, including p21CIP/WAF (unpublished data). The majority of the promoters of these genes do not carry canonical E-Box sites, thus suggesting that N-Myc, like c-Myc, may repress their transcription through indirect binding to promoters. To specifically address this problem, we thought of combining the use of formaldehyde with other cross-linking agents to improve the formation of covalent links between proteins and stabilize the association of proteins to promoters, including those that are not directly bound to DNA. A few papers had been recently published describing the efficacy of several cross-linking agents in ChIP to detect binding of the transcription factors to target genes (Kurdistani et al., 2002; Noma et al., 2006; Nowak et al., 2005; Zeng et al., 2006) Based on these studies we set up a dual cross-linking ChIP protocol that we have successfully employed to improve immunoprecipitation of complexes in which tested factors are not in direct contact with DNA. Specifically, dual cross-linking ChIP was used to check whether N-Myc may associate with the p21CIP/WAF promoter to repress transcription. In Figure 1 we compare results obtained using standard ChIP or the dual cross-linking ChIP. As it can be observed in the Figure, dual cross-linking ChIP significantly improves recovery of p21CIP/WAF promoter and confirms the association of N-Myc to it. Importantly, the sequential use of two cross-linking agents does not affect the recovery of other DNA regions bound by N-Myc directly (see ChIP results for the Apex-1 gene promoter).
In conclusion the protocol described below, can be used to improve in vivo detection of proteins that do not contact DNA directly, though being part of chromatin. Moreover, results suggest that the use of novel cross-linking agents may help increase ChIP sensitivity.
Procedure
The step by step protocol is described for cultured cells grown in two 100mm dishes, containing 1-1.5×107 cells per dish. Two 100mm dishes are used for each immunoprecipitation. In the specific case the protocol is intended for human neuroblastoma cells growing adhesively. Minor adjustments have to be introduced for other cell types especially for those growing in suspension. Based on our experience, one of the most critical steps in performing ChIP regards the conditions of chromatin fragmentation, which need to be empirically set up for each cell type employed.
- Remove medium and add 2ml PBS 1X/1mM PMSF to each plate and scrape cells at room temperature (RT). Pool together the cells from two plates and centrifuge at 1500 rpm for 5 minutes at RT;
- Wash cell pellet with 20ml PBS1X/1mM PMSF at RT and centrifuge at 1500 rpm for 5 minutes. Repeat this step 3 times;
- Resuspend pellet in 20ml PBS1X/1mM PMSF;
- Add disuccinimidyl glutarate (DSG) to a final concentration of 2mM and mix immediately. DSG is prepared as a 0.5M stock solution in DMSO (note 1);
- Incubate for 45 minutes at RT on a rotating wheel at medium speed (8-10 rpm);
- At the end of fixation, centrifuge the sample at 1500 rpm for 10 minutes at RT;
- Wash cell pellet with 20ml PBS1X/1mM PMSF at RT and centrifuge at 1500 rpm for 5 minutes. Repeat this step 3 times;
- Resuspend pellet in 20ml PBS1X/1mM PMSF;
- Add 540µl of 37% formaldehyde and mix immediately. Incubate samples on a rotating wheel for 15 minutes at RT;
- Add 1ml glycine from a 2.5M stock solution and mix immediately. Incubate on a rotating wheel for 10 minutes at RT (note 2);
- Centrifuge samples at 1500 rpm for 4 minutes in cold centrifuge, then keep samples on ice;
- Remove the supernatant and wash pellet 3 times with 10ml ice-cold PBS1X/1mM PMSF. After each washing centrifuge at 1500 rpm for 5 minutes at 4°C;
- Remove supernatant and resuspend pellet in 500µl ice-cold Cell Lysis Buffer. Pipette up and down 10-20 times, then incubate on ice for 10 minutes;
- Centrifuge at 3000 rpm for 5 minutes at 4°C;
- Remove supernatant and resuspend pellet in 600µl ice-cold RIPA Buffer. Pipette up and down 10-20 times, then incubate on ice for 10 minutes;
- Sonication of cross-linked cells is performed in two distinct steps. First, cells are sonicated with a Branson digital Sonifier 2 times for 30 seconds at 40% setting. Next, cell samples are further sonicated with the Diogene Bioruptor for 20 minutes at a full power in a tank filled with ice/water in order to keep cell samples at low temperature during sonication (note 3);
- Centrifuge samples at 14000 rpm for 15 minutes at 4°C;
- Transfer supernatant to a new tube and pre-clear lysate by incubating it with 50µl of Immobilized Protein A (Pierce) for 15 minutes in the cold room at constant rotation;
- Centrifuge samples at 3000 rpm for 5 minutes at 4°C;
- Take the supernatant, after having saved 50µl aliquot for preparation of Input DNA, and add 5µg of specific antibody;
- Rotate the sample O/N in the cold room;
- Add 50µl of Immobilized Protein A (Pierce) (prepared as described in note 4), and incubate by constant rotation for 30 minutes at room temperature;
- Centrifuge the sample at 4000 rpm for 5 minutes at room temperature;
- Remove the supernatant and proceed to wash the beads. For each wash, incubate the sample by constant rotation for 3 minutes at room temperature and the centrifuge at 4000 rpm for 2 minutes at RT;
- Wash 4 times with 1ml RIPA Buffer + Complete protease inhibitors (Roche);
- Wash 4 times with 1ml Washing buffer + Complete protease inhibitors (Roche);
- Wash 2 times with 1ml TE buffer + Complete protease inhibitors (Roche);
- Remove the supernatant and add 200µl TE buffer to the beads. Add 10µg RNAse A and incubate at 37°C for 30 minutes. RNAse was prepared as a 1mg/ml stock solution in TE;
- Add 50µl Proteinase K Buffer 5X and 6µl Proteinase K (19mg/ml). Then, incubate at 65°C in a shaker at 950 rpm for 6 hours;
- Centrifuge at 14000 rpm for 10 minutes at 4°C, then transfer the supernatant (250µl) to a new tube;
- Extract once with phenol/chloroform/isoamyl-alcohol;
- Recover the aqueous phase (200µl) and transfer to a new tube. Add 100µl TE buffer to the remaining phenol/chloroform fraction and re-extract DNA. Recover the aqueous phase and add it to the previous one;
- Extract the recovered aqueous phase once with chloroform/isoamyl-alcohol and transfer to a new tube;
- Add 1µl glycogen (glycogen is 20mg/ml stock solution), 10µg Salmon Sperm DNA, 1/10 volumes Na-acetate 3M pH 5.2, and 2.5 volumes of cold 100% ethanol;
- Vortex and precipitate at -80°C for 40 minutes;
- Centrifuge at 14000 rpm for 30 minutes at 4°C;
- Remove the supernatant and wash pellet with 200µl of cold 70% ethanol;
- Resuspend IP-DNA and INPUT samples in 50-100µl 10mM TrisHCl pH 8;
- Use 2-4µl of IP-DNA and 2µl of Input DNA for Real Time PCR analysis.
Materials & Reagents
| Cell Lysis Buffer | 5mM PIPES pH 8 85mM KCl 0.5% NP40 1mM PMSF Protease inhibitor cocktail (Roche) |
| RIPA Buffer | 150mM NaCl 1% NP40 0.5% NaDoc 0.1% SDS 50mM TrisHCl pH 8 1mM PMSF Protease inhibitor cocktail (Roche) |
| Washing buffer | 100mM TrisHCl pH 8 500mM LiCl 1% NP40 1% NaDoc Protease inhibitor cocktail (Roche) |
| DSG | Di(N-succinimidyl) glutarate Cat 80424 BioChemika, 97.0% (CHN) (Fluka) |
| EGS | Ethylene glycol disuccinate bis(sulfo-N-succinimidyl) ester Cat 39104, BioChemika, puriss., for protein modification, ≥98.5% |
| DMA | Dimethyl adipimidate dihydrochloride Cat 285625, 99% (Aldrich) |
| DSS | Suberic acid bis(N-hydroxysuccinimide ester) (1) Cat S1885, 95%, powder (Sigma) |
Author Notes
- We have tested several crosslinking agents including DSG, EGS [ethylene glycol bis(succinimidylsuccinate], DMA and DSS. In our conditions, we could not observe any improvement of the technique when using DMA and DSS. On the contrary, both DSG and EGS significantly improved the ChIP assay, although DSG worked better than ESG
Sometimes, insoluble aggregates form when DSG is added to cells resuspended in PBS 1X. However, we found that this does not affect the efficiency of the cross-linking reaction. - We believe that it is critical to follow the order of how cross-linkers are added to cells (first DSG and then Formaldehyde) as described in the protocol. In fact, in the reverse order after formaldehyde has worked for the proper time, formaldehyde needs to be quenched out with a certain volume of 2.5M glycine before proceeding with the following crosslinker otherwise it will work too much and chromatin will become resistant to sonication. Unfortunately, based on our experience, no matter how extensively you wash out glycine from cells, even small amount of it can affect DSG activity, thus compromising the reproducibility of the protocol. On the contrary in our conditions glycine is added at the very end of the crosslinking procedure and from that point on whether glycine be perfectly removed or not from cells becomes irrelevant for the following steps.
- Through this procedure we could efficiently fragment neuroblastoma cell chromatin in a range between 500 and 200 bp. As stated above, this is a critical step that must be empirically set up for each cell line tested. For example, HL-60 cells, that grow in suspension, are sonicated with a Branson Sonifier 4 times for 30 seconds at 40% setting and subsequently with the Biogene Bioruptor at a full power for 30 minutes. This procedure allows fragmentation of HL-60 cell chromatin to a size range of 500-1000 bp. Please note that use of two crosslinking agents can make chromatin more resistant to sonication. Therefore based on the type of cells employed it is sometimes necessary to increase the time or power of sonication. As an example please see Figure 2 in which, we compare the average size of DNA fragments obtained from HL-60 cells treated with Formaldehyde alone or DSG + Formaldehyde but using the same conditions of sonication. As you may see from the picture, the combined use of the two cross-linkers make cells more resistant to fragmentation by sonication.
- Preparation of immobilized protein A beads. 500µl of 50% slurry immobilized protein A beads were centrifuged at 2000 rpm for 1 minute. Beads were washed three times with 1ml RIPA Buffer and finally resuspended in 1ml RIPA Buffer. 50 µg Salmon Sperm DNA and 50µg BSA (PCR grade) were added to the beads to allow coating. Beads were incubated for at least 4 hours on a rotating wheel at 4°C. Then, they were centrifuged and washed three times with RIPA Buffer. Finally they were resupended in 500µl RIPA Buffer and ready to be used.
Reviewer Comments
Reviewed by: Bernd Schuettengruber, Cavalli Lab, Institute of Human Genetics, CNRS, Montpellier, France
- Steps 1-12:
- We don’t use PMSF. It’s very unstable in aqueous phase and according to our experience it’s not necessary. One may thus skip it altogether.
- For cells that are tightly attached to the cell culture plate, we propose to perform cross-linking directly on the plates, followed by 2 washing steps with PBS. This would help speeding up the protocol. - Step 15: In our hands longer incubation of lysed cells (30 minutes to 1 hour) on ice before sonication increases the reproducibility of sonication efficiency.
- Step 17: According to our experience, in order to get rid of all the cell debris it is more efficient to centrifuge 2 times (for 5 minutes) rather than once for 15 minutes.
- Step 18: We pre-clear at least 2 hours or overnight. This may help to reduce background.
- Steps 21 and 22: We add antibodies for 4 hours (during the day), and then again incubate the samples with beads overnight.
- Steps 32 and 33: We only do one phenol/chloroform/isoamyl-alcohol extraction. The re-extraction might be useful when working with very low amounts of chromatin.
Figures
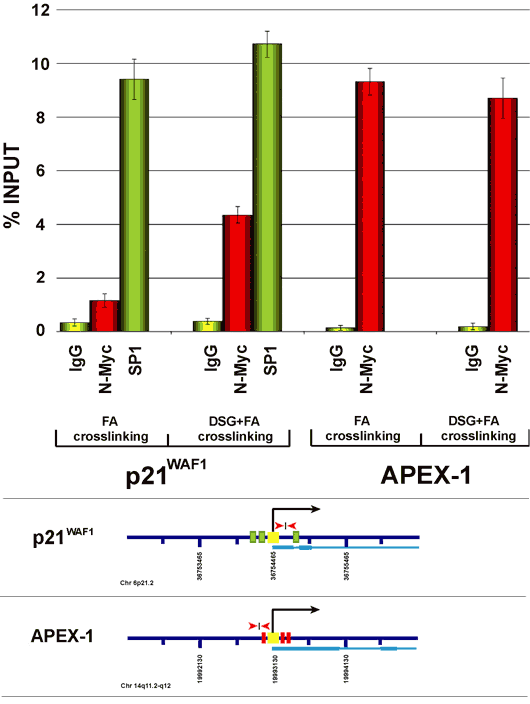
Figure 1: ChIP analysis of the p21CIP/WAF and APEX-1 promoters. Maps of gene promoters contain: transcription start site (black arrow and yellow box); Sp1 binding sites (green boxes); Myc/Max binding sites (red boxes); DNA regions amplified by qChIP (red arrows); exon-intron structure (light blue). ChIP results are expressed through histogram representations. Enrichment of a specific DNA region has been quantified as a percent recovery of the input. Histogram values are the average of three independent ChIP experiments in which quantitative Real Time PCR has been performed in triplicate each time (error standard deviation is included).

Figure 2: Average size of DNA fragments obtained from H-60 cells treated with one or two cross-linkers. DNA was separated on a 1.5% agarose gel, TBE1X. 1kb ladder has been used as a marker of DNA size. FA:Formaldehyde, DSG: disuccinimidyl glutarate.
References
- Gartel, A.L., Ye, X., Goufman, E., Shianov, P., Hay, N., Najmabadi, F., and Tyner A.L. (2001) Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3, Proc Natl Acad Sci USA, 98: 4510-4515.
- Kurdistani, S.K., Robyr, D., Tavazoie, S., and Grunstein, M. (2002) Genome-wide binding map of the histone deacetylase Rpd3 in yeast, Nature Genet., 31: 248-254.
- Noma, K., Cam, H.P., Maraia, R.J., and Grewal S.I.S. (2006) A role for TFIIIC Transcription Factor Complex in Genome Organization, Cell, 125: 859-872.
- Nowak, D.E., Tian, B. and Brasier, A.R. (2005) Two-step cross-linking method for identification of NF-kB gene network by chromatin immunoprecipitation, Biotechniques, 39: 715-725.
- Perini, G., Diolaiti, D., Porro, A. and Della Valle, G. (2005) In vivo transcriptional regulation of N-Myc target genes is controlled by E-box methylation, Proc Natl Acad Sci USA, 102: 12117-12122.
- Wu, S., Cetinkaya, C., Munoz-Alonso, M.J., von der Lehr, N., Baharam, F., Beuger, V., Eilers, M., Leon, J. and Larsson L.G. (2003) Myc represses differentiation-induced p21CIP1 via Miz-1-dependent interaction with the p21 core promoter, Oncogene, 22: 351-360.
- Zeng, P-Y., Vakoc, C.R., Chen, Z-C., Blobel, G.A. and Berger S.L. (2006) In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation, Biotechniques, 41: 694-698.

